MultiPsi: Quantum Chemistry on HPC for Photochemistry and Inorganic Chemistry
Mickael Delcey, Division of Computational Chemistry, Lund University
Modelling Chemistry
Computational chemistry has revolutionised science and industry and helped to improve our understanding of chemical processes. Instead of relying solely on trial and error in the lab, researchers can now predict how molecules will behave under different conditions using computer simulations, thereby accelerating, for example, drug design or the development of materials for applications ranging from electronics to energy storage. These advancements have only been made possible by simultaneous developments of new computational chemistry methods, more powerful computers and more efficient implementations that leverage the power of emerging computer architectures. Thus, while the first quantum descriptions of atoms and simple molecules date back a hundred years, it is only in recent decades that quantum chemistry has become a realistic tool for industry.
One Size Does Not Fit All
Quantum chemistry encompasses a broad collection of methods, each with its own strengths and weaknesses. Some of this diversity simply corresponds to increasingly sophisticated approximations that yield better results at a higher computational cost. However, the choice of an appropriate method often depends on the specific system or property of interest. What works well for one molecule or chemical property may fail dramatically in another case. One important example is the approximation that the electronic structure of a molecule is well described by a single electron configuration. This is the case for most organic molecules: molecules composed mainly of carbon, hydrogen, oxygen and nitrogen, which are prevalent in living beings. In these cases, computationally inexpensive methods, like perturbation theory or density functional theory (DFT), are very effective. However, these approximations sometimes break down, in particular for molecules containing transition metals like iron, manganese, or chromium. Yet these atoms are crucial in solid-state materials and also in biological systems. Indeed, even if they represent a small fraction of the elements in living beings, transition metals are estimated to be present in a third of all proteins and enzymes where they are often key to their roles. Transition metals are, in particular, essential to some of the most important bioreactions, such as photosynthesis or nitrogen fixation (a reaction in some bacteria which contributes to soil fertilisation). A similar breakdown of these approximations can occur in photochemistry, for example, at the critical moment when an excited molecule relaxes back into its ground state without emitting light. In these cases, the correct wave function can be built using a (hopefully small) number of configurations, giving rise to so-called multiconfigurational methods.
Modularity
The abundance of quantum chemistry methods has led to the implementation of numerous algorithms in most quantum chemistry software. Consequently, these programs become large and unwieldy, posing challenges for long-term maintenance and hindering accessibility for new students and researchers. To address this issue, there is a growing push towards developing small modular programs that excel at specific tasks, promoting consistency and interoperability [4]. This shift aims to streamline code development and usage, thus reducing redundancy and facilitating collaboration within the research community.
Some years ago, development of the VeloxChem program [6] started at the KTH Royal Institute of Technology (KTH), with active support from PDC staff and promised a highly efficient code for all types of chemical properties at the Hartree-Fock and DFT level. This initiative paid off, and some of the code’s impressive performances have been reported in previous PDC newsletters. This world-leading development inspired a number of groups, including ours, to develop additional libraries covering alternative electronic structure methods. Thus, while VeloxChem covers Hartree-Fock and DFT (the most inexpensive ab initio quantum chemistry methods), Gator [5], which was developed in partnership with Heidelberg University, expands the functionality with the algebraic diagrammatic construction (ADC) method, which is a more expensive but reliable wave function method for organic molecules. Similarly, the MultiPsi [1] library, which was developed mostly at Lund University, expands the functionality towards multiconfigurational methods to describe, among others, metal complexes.
Goals and Ambitions for MultiPsi
Multiconfigurational methods can often be thought of as generalisations of standard single configurational methods and have, in principle, a computational cost that is only moderately higher. Yet, they have received less attention and, for this reason, lag behind in efficiency. One of the main aims of MultiPsi is to enable truly large-scale multiconfigurational calculations and, in particular, to be fully adapted to modern heterogeneous parallel computer architectures. The program should be, for instance, efficient enough to simulate large metalloproteins or the dynamics of medium-sized molecules after light absorption for nanoseconds or longer.
But beyond this, these methods are also known to be more challenging to use and develop and are rarely presented in great detail in standard textbooks. For this reason, education is a significant aspect of making these methods more widely used and promoting further developments. MultiPsi was thus designed with usability as a fundamental property as well as high interactivity. With this mindset, along with the rest of the VeloxChem community, we have set up a collection of Jupyter notebooks forming an e-book meant for teaching quantum chemistry at all levels, which we call the e-Chem initiative [2].
Finally, MultiPsi is not meant to simply implement existing methods in an efficient way but to be a development platform for modern multiconfigurational quantum chemistry. Its structure should make it easy to develop new code, from early prototypes to final implementations, even for new students who have limited prior programming experience.
Structure of MultiPsi
In practice, MultiPsi, like all of our programs, has a two-layer programming structure. The upper layer, written in Python, is where most of the quantum chemical methods are implemented, and it also manages the use of the available hardware resources using message passing interface (MPI) communicators. The lower layer, written in C++, contains the compute-intensive core functionalities. Finally, the code uses a number of external libraries, including VeloxChem.
We can illustrate this structure for the multiconfigurational self-consistent field method (MCSCF), which is one of the fundamental multiconfigurational methods in much the same way as the Hartree-Fock method is for single configurational methods. The MCSCF algorithm is implemented in Python but requires two computationally intensive functionalities: the evaluation of the electron repulsion integrals (ERIs), which represent the interactions between electrons, and the configuration interaction (CI) to evaluate the relative weights of all configurations. The ERIs are a central quantity in all of quantum chemistry, as they allow us to transform the differential equations of quantum mechanics into a linear algebra computer program. They are expensive and numerous, scaling as N4 with the system size, though efficient screening methods can reduce this scaling significantly. Their evaluation is performed by C++ routines in VeloxChem, but, through Pybind11, these routines are exposed to the Python layer and can then easily be used by MultiPsi by importing the VeloxChem module. The CI functionalities are, on the other hand, exclusive to MultiPsi and implemented in their own C++ layer. The overall structure can be seen in the figure below.
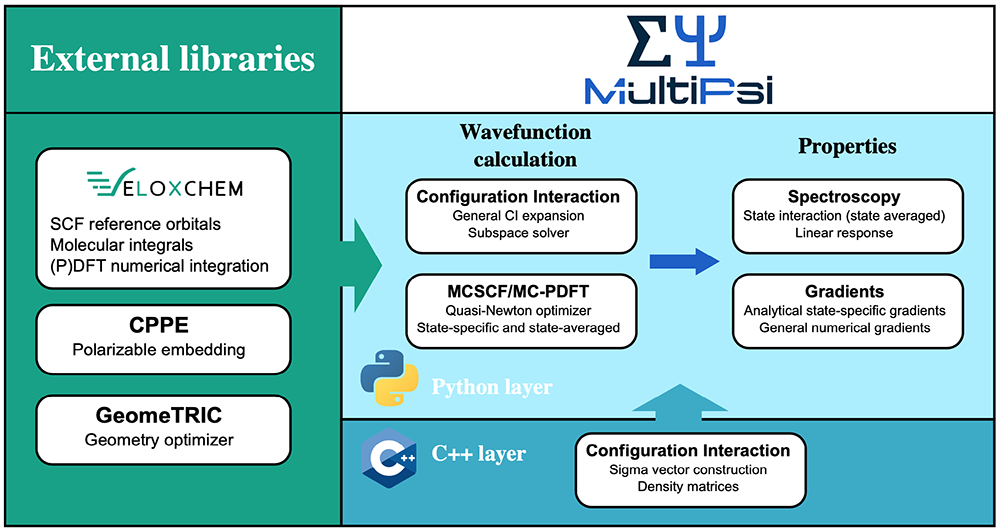
The modularity of the design means, however, that every part can easily be substituted. For example, MultiPsi has been used in a prototype of hybrid quantum computing code [3] by substituting its CI module with the module QiSkit from IBM, which is able to perform similar function but on a quantum computer. Writing this prototype took less than a day for a student who was not familiar with MultiPsi’s code prior to doing this!
Efficiency
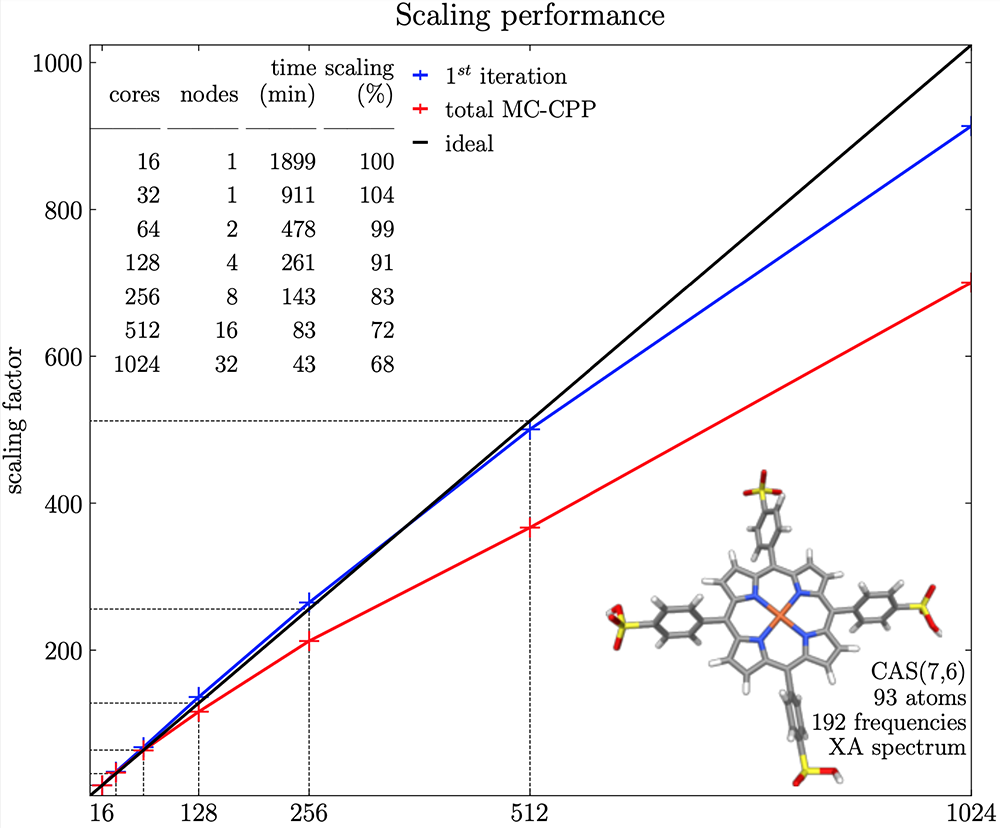
MultiPsi has been written from scratch around heterogeneous parallel computer architectures. Taking MCSCF as an example again, as discussed above, the cost is essentially the sum of an ERI-driven part and a CI part. By using the general ERI evaluator from VeloxChem, MultiPsi inherits its efficiency with only a little work being needed to control the task distribution. This gives the parallel performance shown in the figure below. This figure shows both the parallel efficiency for the first iteration and for the total calculation. The timing for the first iteration is almost exclusively made up of the ERI evaluation, while the (partly single-node) convergence algorithm starts to dominate for a large number of nodes due to Amdahl’s law.
The second part of the cost is the CI, which depends on the number of configurations. Configurations are typically chosen by allowing all possible distributions of a subset of electrons in a subset of orbitals in the system. Due to the combinatorial nature of this technique, the number of configurations grows very fast (factorially) with the number of electrons and orbitals. As a result, while the most expensive operation in a CI calculation is a simple matrix-vector product, the size of the vector (the number of configurations) can quickly reach billions of double-precision numbers. For such sizes, the CI matrix is completely impossible to store, even taking advantage of its significant sparseness. Instead, it is computed on the fly and immediately multiplied by the CI vector. The choice of algorithm then becomes a fine balance between vectorisation and maximal utilisation of the sparseness.
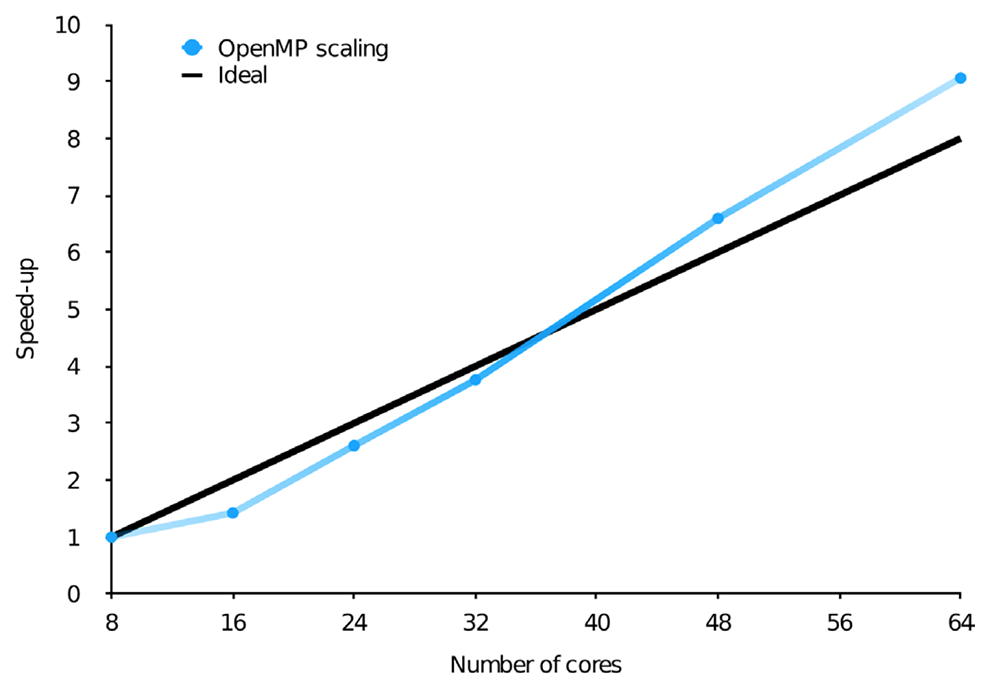
Still, with billions of configurations, even the CI vector can become a memory bottleneck. Thus, when designing parallel algorithms, it is essential for these vectors to be stored in the distributed memory. This, however, necessarily leads to high inter-node communication since in a matrix-vector multiplication, in principle, each resulting vector element has contributions from all the starting vector elements. This can be greatly reduced by making use of the predictable sparsity in the CI matrix, which introduces some level of locality in the data. The resulting OpenMP and MPI parallel efficiency using Dardel are shown in the figures below on the top (a) and bottom (b), respectively.
Scaling of the wall time of the CI step in two different calculations, with the number of OpenMP threads or MPI ranks

In the end, this efficiency has allowed MultiPsi to shatter several records. In our release paper, we broke the record for the largest CI optimisation. Using 128 nodes on the Dardel system at PDC, we optimised the CI weights for more than 400 billion configurations. At this size, a single CI vector is about 3TB of distributed memory, and a couple of them need to be stored at a given time. In addition, we also significantly pushed the limit of the largest molecule ever computed by MCSCF. We have now demonstrated MultiPsi’s ability to simulate a biomolecule of nearly 1,500 atoms using only 256 cores [8]. It is generally considered that 1,000 atoms are sufficient for a quantum chemical calculation, as atoms far apart from the region of interest can then be advantageously modelled using significantly cheaper classical molecular mechanics – a form of multiscale modelling. With these performances, MultiPsi is arguably the most efficient platform for multiconfigurational calculations in the world.
Outlook: Towards New Science
MultiPsi is already available through Conda for testing and is going to be released officially under the GNU Lesser General Public License version 2.1 (LGPLv2.1) sometime this year. Currently, it offers mostly energy and properties (in particular spectroscopy simulations) with the MCSCF method. However, as discussed earlier, this is not enough. Multiconfigurational methods have also lagged behind in functionalities and method availability and require new developments.
In particular, while DFT has been the most used method in quantum chemistry for decades, there has not been a satisfying multiconfigurational variant, despite decades of development. We believe we have finally developed such variant, a true generalisation of DFT to many configurations [8]. We showed that the method indeed extends the accuracy of DFT to systems requiring multiconfigurational methods. In addition, as can be seen in the figure on the next page, for large molecules, this method can be as efficient as DFT, despite it providing a better description of the multiconfigurational nature of the metal centre. This record performance is also thanks to help from a PDC software specialist to parallelise the (P)DFT numerical integration.
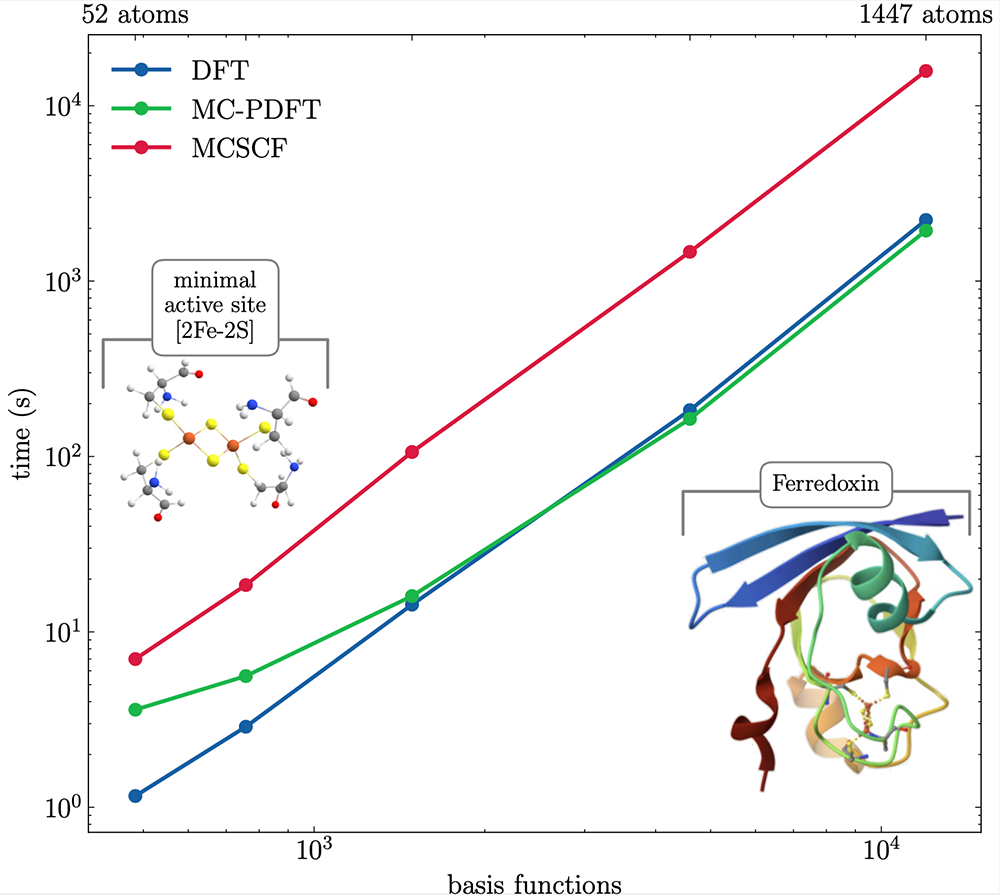
Developments are ongoing to establish this new method and implement property calculations with it. With this, MultiPsi will finally offer quantum chemists a realistic alternative to DFT for metal complexes and other molecules where the latter may not be accurate enough.
Acknowledgements
The VeloxChem and MultiPsi projects are supported by a collaboration with the PDC Center for High Performance Computing. The results presented in this article were obtained with the use of computational resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS).
References
- M. G. Delcey, “MultiPsi: A python-driven MCSCF program for photochemistry and spectroscopy simulations on modern HPC environments”. WIRES Comput. Mol. Sci. 13.6 (2023), e1675. doi.org/10.1002/wcms.1675
- T. Fransson et al., “eChem: A Notebook Exploration of Quantum Chemistry”. In: Journal of Chemical Education” 100.4 (2023), pp. 1664–1671. doi.org/10.1021/acs.jchemed.2c01103
- J. de Gracia Triviñ̃o, M.G. Delcey, G. Wendin, “Complete Active Space Methods for NISQ Devices: The Importance of Canonical Orbital Optimization for Accuracy and Noise Resilience”. Journal of Chemical Theory and Computation 19.10 (2023). PMID: 37103120, pp. 2863–2872. doi.org/10.1021/acs.jctc.3c00123
- S. Lehtola, “A call to arms: Making the case for more reusable libraries”. The Journal of Chemical Physics 159.18 (Nov. 2023), p. 180901. issn: 0021-9606. doi.org/10.1063/5.0175165
- D. R. Rehn et al., “Gator: A Python-driven program for spectroscopy simulations using correlated wave functions”, WIREs Comput. Mol. Sci. 11.6 (2021), e1528. doi.org/10.1002/wcms.1528
- Z. Rinkevicius et al., “VeloxChem: A Python-driven density-functional theory program for spectroscopy simulations in high-performance com- puting environments”. WIREs Comput. Mol. Sci. 2020, 10:e1457. doi.org/10.1002/wcms.1457
- M. Scott, M. G. Delcey, “Complex Linear Response Functions for a Multiconfigurational Self-Consistent Field Wave Function in a High Performance Computing Environment”. Journal of Chemical Theory and Computation 19.17 (2023), PMID: 37596971, pp. 5924–5937. doi.org/10.1021/acs.jctc.3c00317
- M. Scott et al., “Variational Pair-Density Functional Theory: Dealing with Strong Correlation at the Protein Scale”. Journal of Chemical Theory and Computation 20.6 (2024), PMID: 38217859, pp. 2423–2432. doi.org/10.1021/acs.jctc.3c01240